Research
Our research lies at the intersection of artificial intelligence, applied mathematics, network science, and computational biology. We develop computational and data-driven approaches to study cellular dynamics, genomic regulation, neurological disease, complex biological networks, and high-dimensional biomedical data.
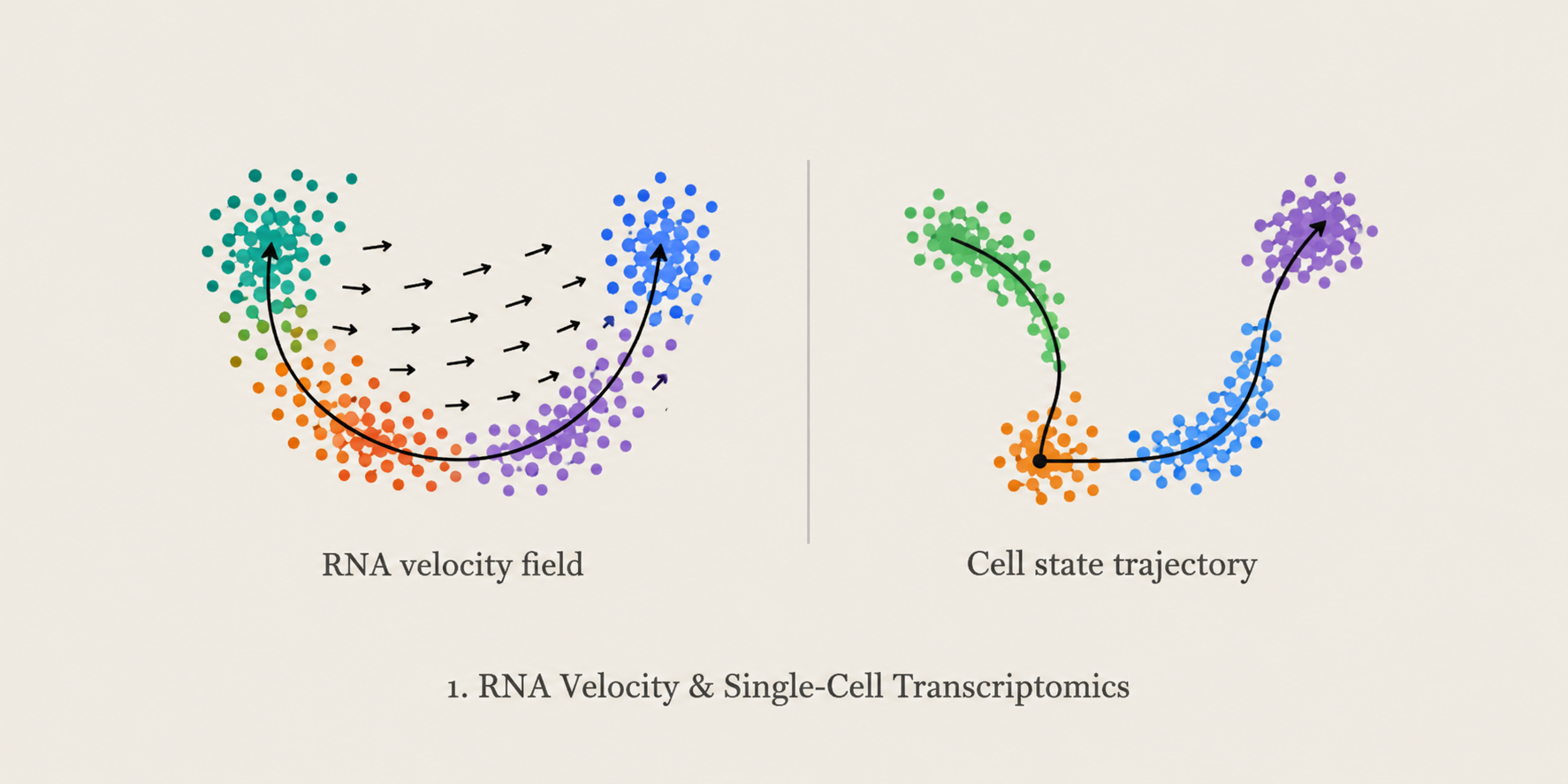
RNA Velocity & Single-Cell Transcriptomics
We develop computational and topology-aware approaches to study cellular dynamics and developmental trajectories using single-cell transcriptomics and RNA velocity. Current work includes EVTS, a framework for evaluating RNA velocity embedding faithfulness, trajectory stability, and the relationship between projected velocity vectors and future cellular states.
Methods: scRNA-seq · RNA Velocity · Topology · EVTS · Trajectory Inference
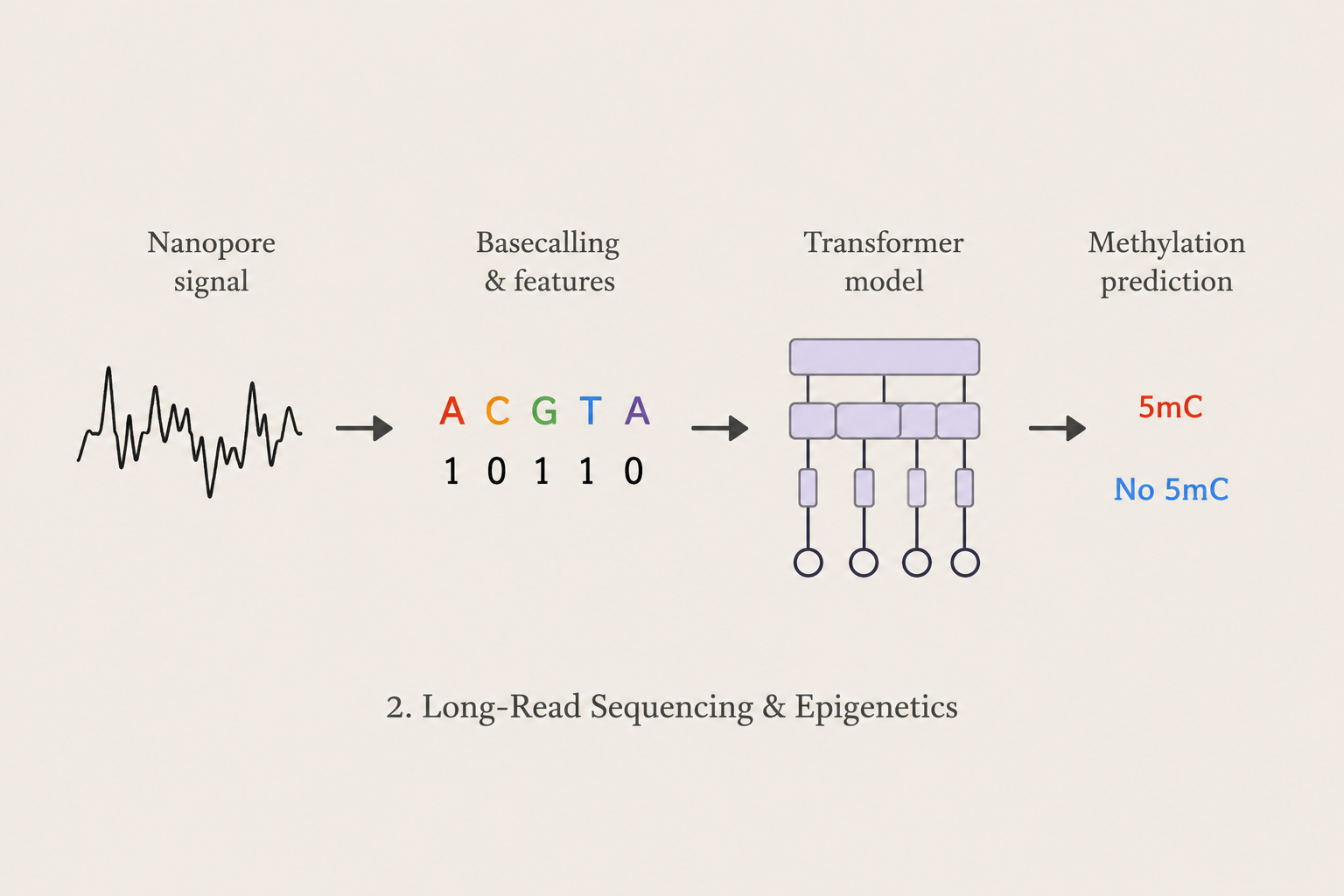
Long-Read Sequencing & Epigenetics
We develop machine learning and mathematical approaches for extracting biological information from Nanopore long-read sequencing data. Our research investigates signal-level DNA methylation detection, epigenetic modifications, isoform-level analysis, and computational methods for long-read genomic and transcriptomic data.
Methods: Nanopore Sequencing · Transformers · Deep Learning · DNA Methylation · Signal Processing
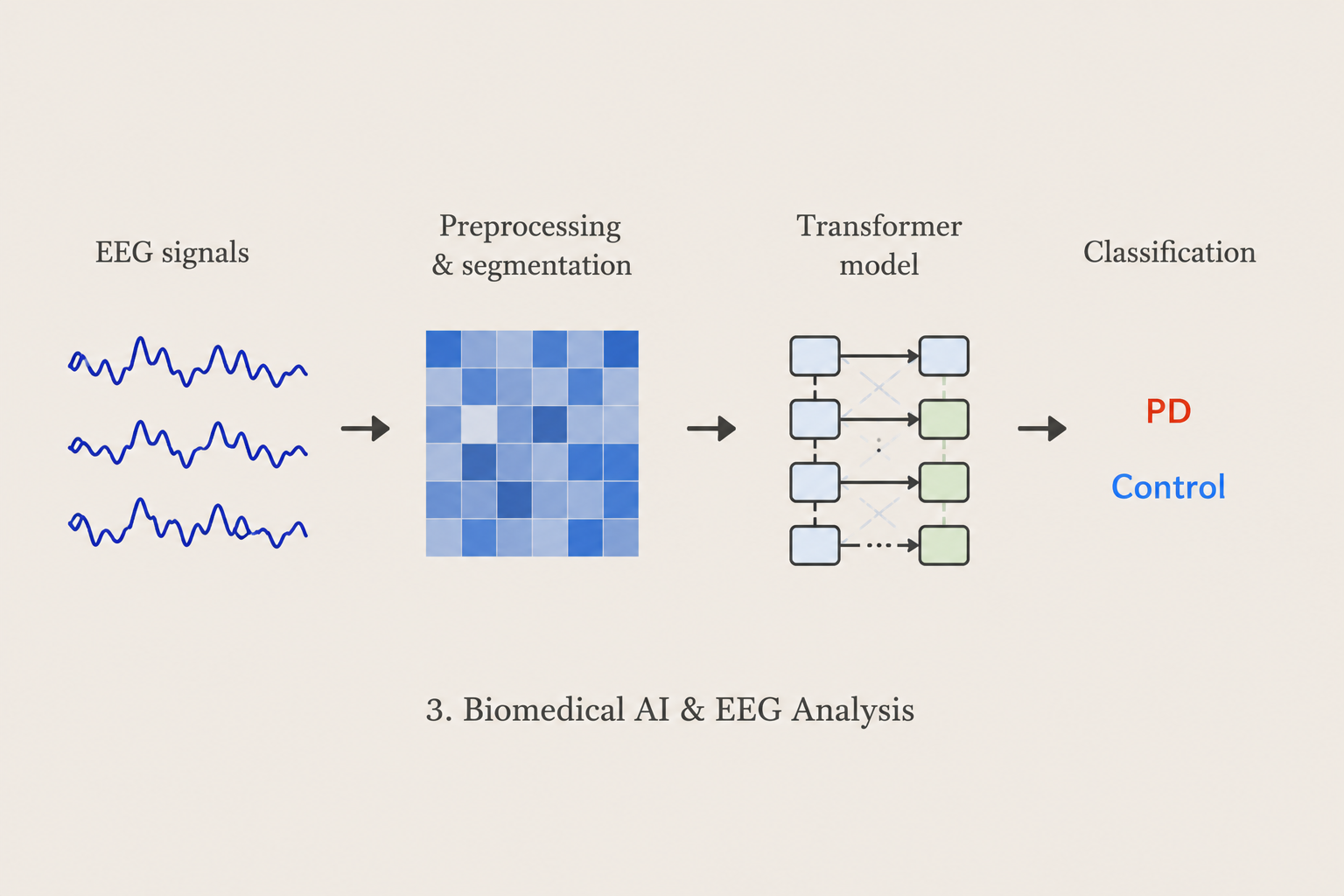
Biomedical AI & EEG Analysis
We investigate artificial intelligence and deep learning methods for biomedical signal analysis. Our work applies Transformer-based architectures to EEG recordings for neurological disease classification, representation learning, and cross-subject prediction, with applications to Parkinson’s disease and other biomedical problems.
Methods: EEG · Transformers · Deep Learning · Classification · Neuroinformatics
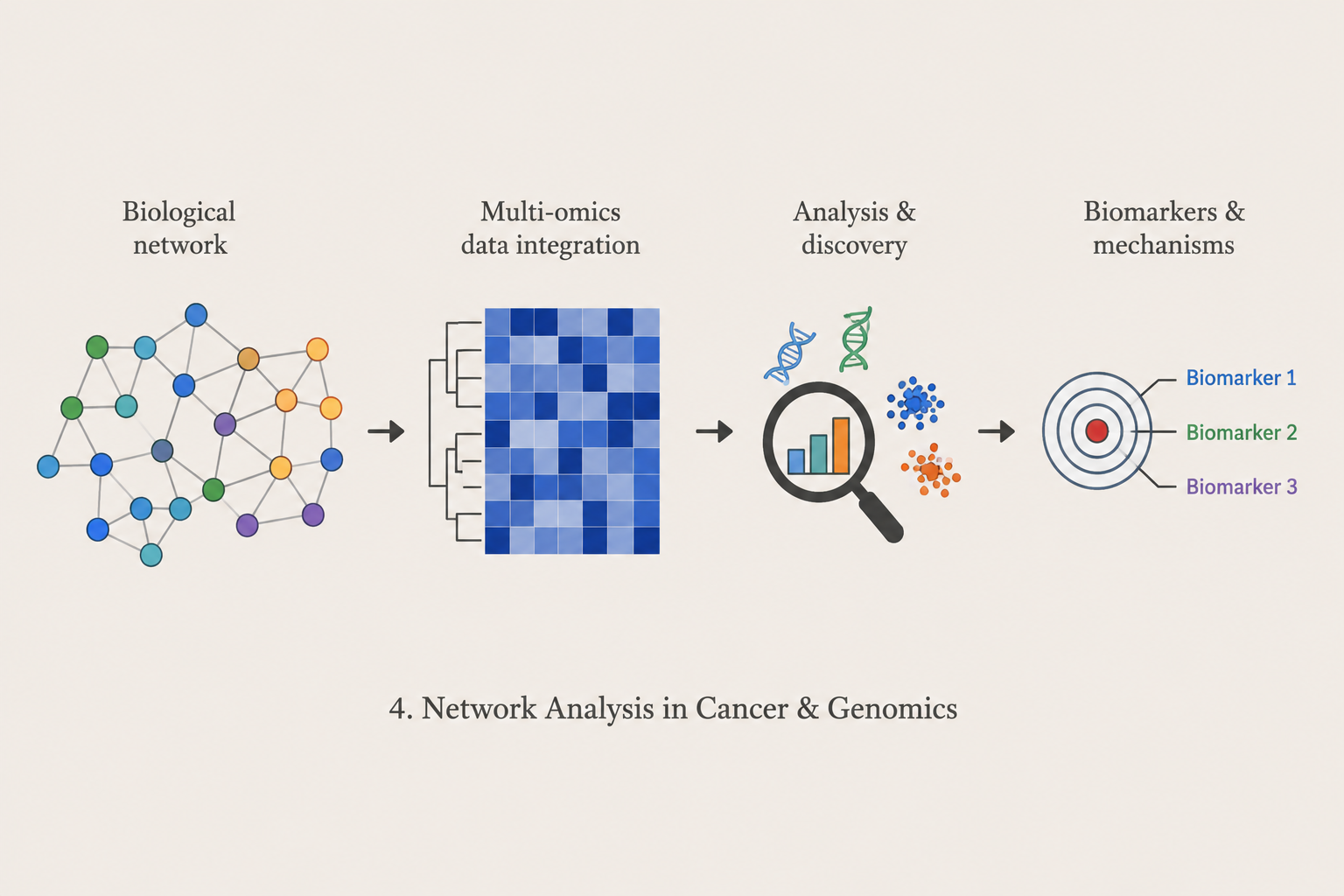
Network Analysis in Cancer & Genomics
We construct and analyze biological networks to investigate molecular organization, transcriptional heterogeneity, and disease mechanisms. Current research includes mega-scale cell-cell similarity networks, community detection, bridge-cell analysis, cancer-associated RNA networks, and macrophage state transitions in non-small cell lung cancer.
Methods: Network Analysis · Community Detection · Graph Analytics · lncRNA · Cancer Bioinformatics
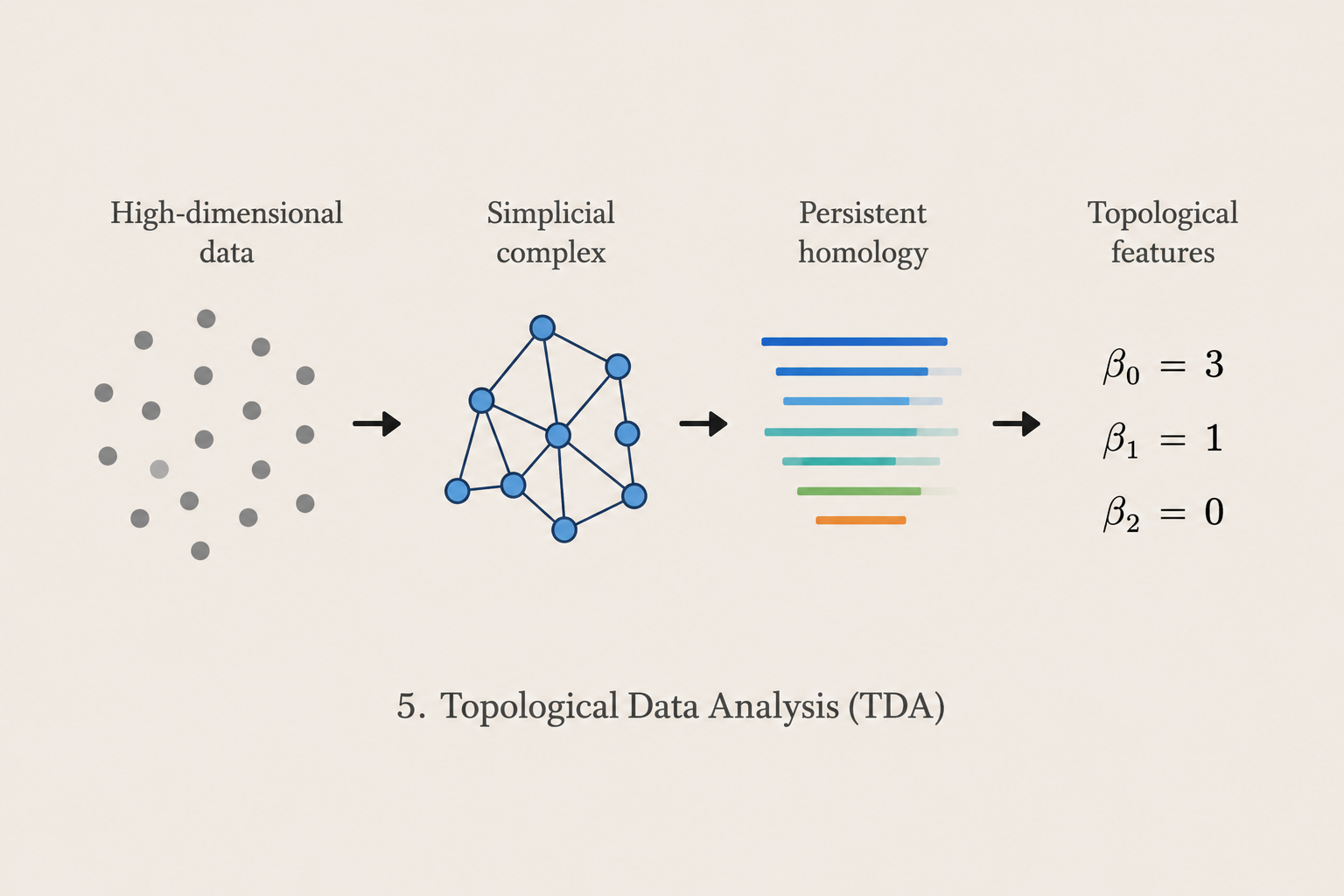
Topological Data Analysis
We apply topological and geometric methods to identify robust structures in complex high-dimensional datasets. Our research investigates topology preservation, persistent features, embedding robustness, and interpretable representations of biomedical and population-level data.
Methods: Topological Data Analysis · Persistent Homology · Mapper · Dimensionality Reduction · Visualization
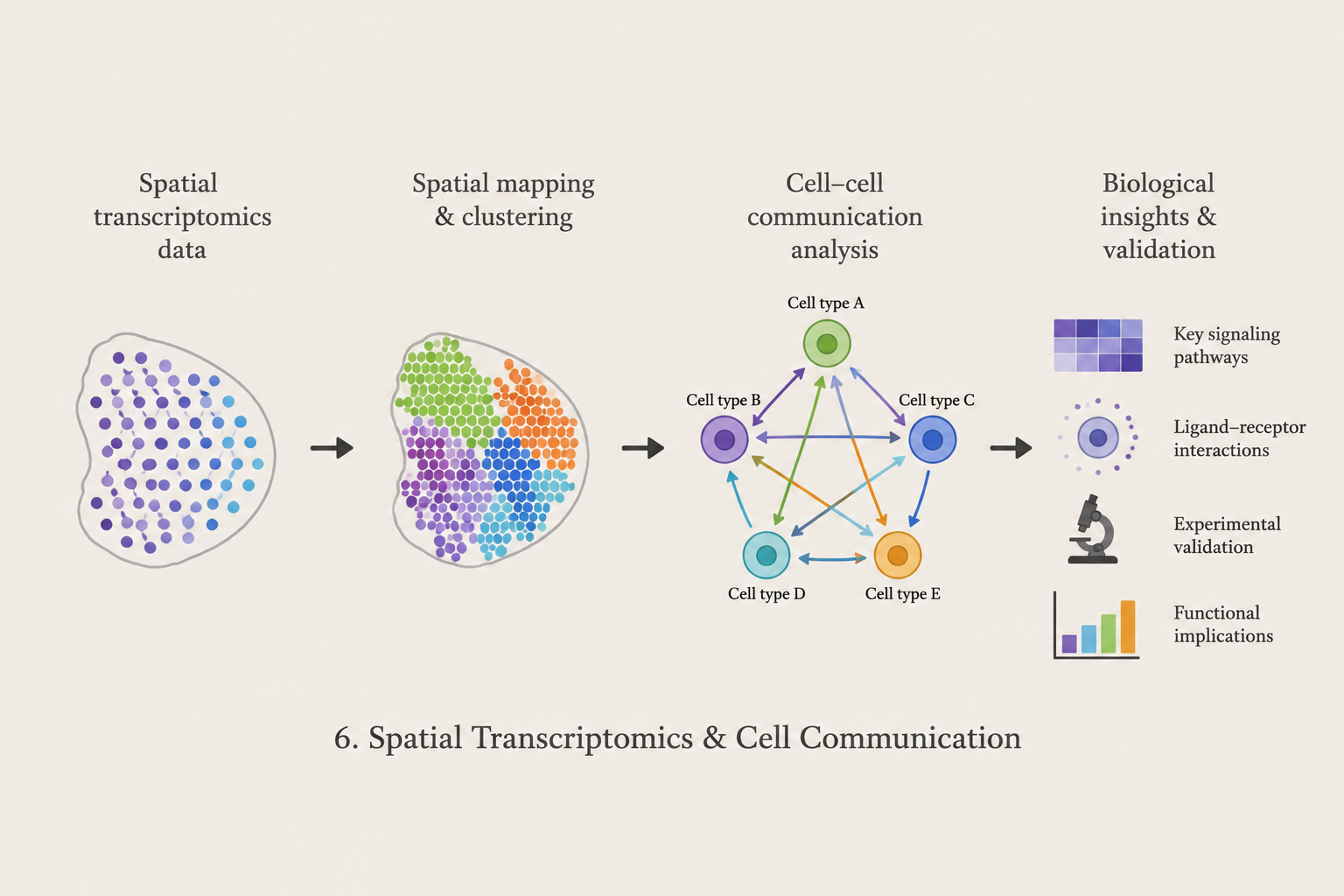
Spatial Transcriptomics & Cell Communication
We are developing computational approaches for analyzing spatially resolved transcriptomic data and cellular interactions within complex biological tissues. This research focuses on spatial organization, cell-cell communication, signaling pathways, and integration with single-cell transcriptomic analysis.
Methods: Spatial Transcriptomics · Cell Communication · Clustering · Ligand-Receptor Analysis · Single-Cell Genomics